
Introduction
RNA sequencing (RNA-seq) has revolutionized transcriptomic analysis, offering detailed insights into gene expression across diverse biological samples. However, the accuracy of these insights is often compromised by batch effects—systematic, non-biological variations introduced during sample processing and sequencing. To overcome these limitations, Xiaoyu Zhang introduces ComBat-ref, a refined method for batch effect correction that builds upon the widely used ComBat-seq approach. By using a reference batch with the lowest dispersion and aligning all other batches to it, ComBat-ref significantly enhances the statistical power of differential expression (DE) analyses while retaining compatibility with popular RNA-seq tools such as DESeq2 and edgeR. This review summarizes the key contributions, methods, data, results, and broader implications of the study.
Contributions of the Paper
– Introduces ComBat-ref, a novel method that refines batch effect correction for RNA-seq count data by using a reference batch strategy.
– Improves upon ComBat-seq by retaining count data from the reference batch while aligning others to match its dispersion profile.
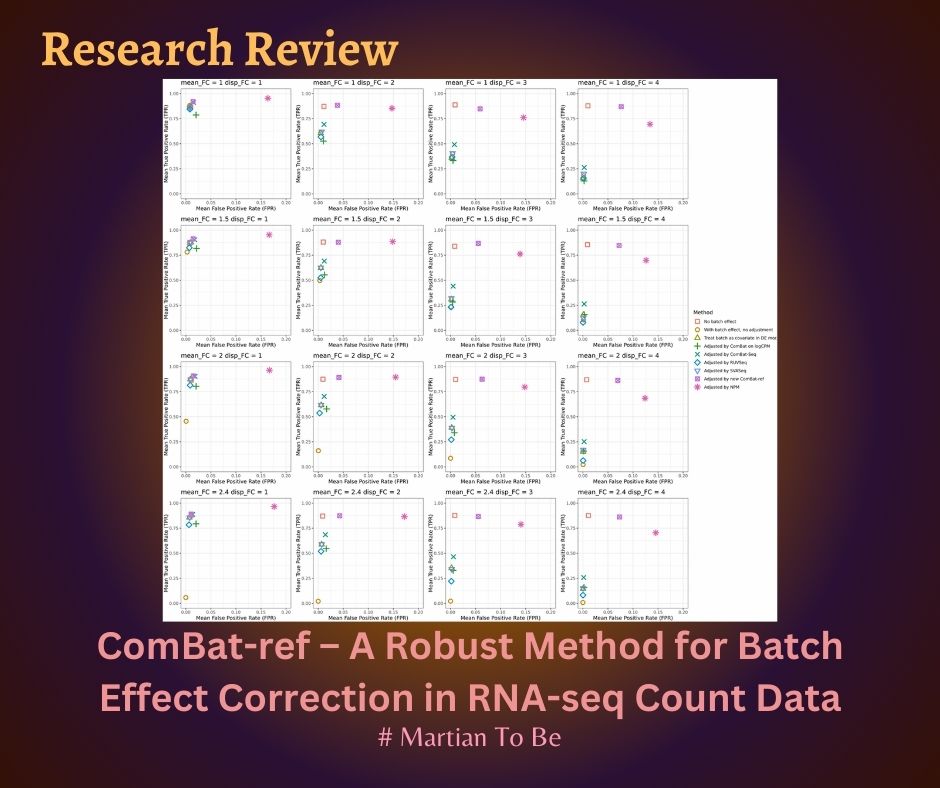
– Demonstrates higher sensitivity and specificity in detecting differentially expressed genes, especially under challenging batch dispersion scenarios.
– Maintains compatibility with integer-count-based DE tools, like DESeq2 and edgeR.
– Successfully applies the method to multiple real-world datasets, showing robustness across diverse experimental designs.
Practical Implications of the Paper
– Enables more accurate gene expression analysis in RNA-seq studies affected by technical batch variations.
– Facilitates the integration of datasets from multiple sources or time points, improving meta-analysis reliability.
– Enhances biomarker discovery by improving detection of true biological signals obscured by batch effects.
– Provides a practical tool for space biology and clinical research, demonstrated through NASA GeneLab and human disease datasets.
Methods Used in the Paper
– Negative binomial modeling for RNA-seq count data, aligning with the statistical assumptions of DESeq2 and edgeR.
– Selection of a reference batch with the lowest dispersion, serving as the baseline for batch adjustment.
– Adjustment of gene counts from other batches using generalized linear models (GLMs) and cumulative distribution function (CDF) matching.
– Comparative benchmarking using simulation studies and real datasets.
Data Used in the Paper
– Simulated datasets generated via the Polyester R package, modeling varying degrees of mean expression and dispersion across batches.
– Growth Factor Receptor Network (GFRN) dataset, involving different oncogenes across batches.
– Human RNA-seq datasets: GSE182440 (Alcohol Use Disorder) and GSE173078 (Periodontal Disease).
– NASA GeneLab transcriptomic datasets, including GLDS_137, GLDS_242, and GLDS_48, which involve complex batch structures from spaceflight experiments.
Results of the Paper
– ComBat-ref consistently outperformed ComBat-seq and NPMatch in both simulations and real data.
– Achieved higher true positive rates (TPR) and lower false positive rates (FPR), especially when dispersion varied greatly among batches.
– Improved clustering accuracy in PCA plots (e.g., Gamma score: 0.66, Dunn index: 1.60).
– Identified more biologically meaningful DE genes, including successful recovery of EGFR with high statistical significance.
– Enhanced pathway enrichment detection, particularly in the RAS signaling pathway.
Conclusions from the Paper
– ComBat-ref provides a reliable, effective method for batch effect correction in RNA-seq studies.
– The reference batch strategy preserves biological variation while minimizing technical noise.
– Demonstrates strong potential for broad adoption in genomics and transcriptomics pipelines.
Limitations of the Paper
– Slightly elevated false positive rate compared to ComBat-seq, although still significantly lower than NPMatch.
– Assumes one dispersion parameter per batch, which may oversimplify more complex batch structures.
– Not yet validated for single-cell RNA-seq (scRNA-seq) datasets with their unique sparsity and heterogeneity challenges.
Future Works Suggested in the Paper
– Adaptation of ComBat-ref for scRNA-seq using sparsity-aware models like zero-inflated negative binomial (ZINB).
– Extension to hierarchical batch models, allowing finer control in multi-level experimental designs.
– Integration with larger scRNA-seq benchmarks to assess performance in single-cell applications.
